Síndrome de Apert
también conocido como: Acrocefalosindactilia.
¿Qué es el Síndrome de Apert?
Síndrome de Apert fue descrito por primera vez en 1894 por el Dr. Wheaton y se caracterizó además por el Dr. Apert en 1906. Esta condición también se conoce como acrocefalosindactilia lo que significa una forma de cúpula de la cabeza con la fusión de los dedos.
¿Qué causa el síndrome de Apert?
Esta condición es causada por un cambio ("mutación") en un gen llamado factor de crecimiento fibroblástico gen del receptor, 2 o más simplemente FGFR2. Cada persona tiene dos copias del gen FGFR2. Sólo se necesita una mutación en uno de los miembros del par de genes FGFR2 a causa de los signos clínicos del síndrome de Apert. El síndrome de Apert se hereda en un patrón de herencia autosómico dominante por lo tanto, si una persona tiene el síndrome de Apert, su / sus hijos tendrían un 50% para tener también el síndrome de Apert.
La mayoría de las personas con síndrome de Apert son los primeros casos en la familia. Esto significa que los padres de los niños afectados no tienen el síndrome de Apert. Si los padres de un niño no tiene el síndrome de Apert, el riesgo para sus otros embarazos es baja. Sin embargo, un mayor riesgo de esta enfermedad se ha encontrado embarazos de padres mayores.
¿Cuáles son los síntomas del síndrome de Apert?
Los principales signos clínicos del síndrome de Apert son los síguientes:
- Craneosinostosis (closuse prematura de las suturas del cráneo) haciendo que la cabeza tiene un alto, "cúpula" apariencia
- Planos del tercio medio facial
- Los ojos muy abiertos el espacio
- El paladar hendido
- Sindactilia de los dedos de manos y pies (fusión de la piel y los huesos de los dedos de manos y pies)
- Otras características del síndrome de Apert incluyen retardo variable del desarrollo, el acné significativo durante la adolescencia, y las anomalías del cerebro.
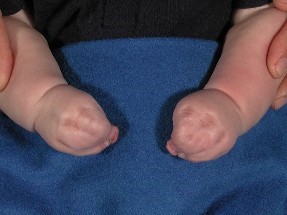
Imagen de manos con sindactilia (fusión de los dedos)
Imagen de manos con sindactilia (fusión de los dedos de los pies)
¿Cuál es el pronóstico para las niños con síndrome de Apert?
Con la atención médica cercana, el pronóstico para la mayoría de los niños con esta afección es positiva. Múltiples cirugías y tratamientos en curso se requieren para estos niños, pero la mayoría en última instancia, tienen una función útil de las manos, ir a la escuela, tener amigos, y sobre todo disfrutar de la vida. Con un seguimiento cercano por un equipo especializado, estos niños pueden crecer a adultos sanos y felices.
Tratamientos y especialistas del Síndrome de Apert
El síndrome de Apert es una afección que dura toda la vida y no tiene cura. Es probable que su hijo necesite consultar a varios especialistas médicos y recibir algunos de los siguientes tratamientos a lo largo de su proceso de atención.
- Cirugía. Un niño puede requerir cirugía poco después del nacimiento si tiene síntomas que afectan el cerebro o el cráneo. Es posible que se necesiten otras cirugías para los ojos, la nariz, el mentón, la mandíbula y los dedos de las manos y los pies.
- Audífonos. Es posible que los especialistas en audición deban recetar audífonos a niños con discapacidad auditiva.
- Terapia. La fisioterapia, la terapia ocupacional y la logopedia son tratamientos potenciales para el síndrome de Apert.
- Máquinas de respiración. Si el niño tiene una obstrucción de las vías respiratorias, es posible que se necesiten especialistas para evaluar las dificultades respiratorias y recetarle un respirador.
- Atención dental. El cuidado de la boca y los dientes del niño suele ser fundamental para los niños con síndrome de Apert.
- Cuidado de la visión. Las evaluaciones periódicas de la visión y el cuidado de la visión también son una parte clave del tratamiento del síndrome de Apert.
Esta página fue actualizada por última vez en: diciembre 15, 2023 04:48 p. m.