Apert Syndrome
Also known as: Acrocephalosyndactyly.
What is Apert Syndrome?
Apert syndrome was first described in 1894 by Dr. Wheaton and was further characterized by Dr. Eugène Charles Apert in 1906. This condition is also called Acrocephalosyndactyly, which means a dome- shaped head with fusion of the fingers.
What causes Apert syndrome?
This condition is caused by a change (“mutation”) in a gene called the Fibroblast Growth Factor Receptor Gene 2 or more simply FGFR2. Each person has two copies of the FGFR2 gene. It only takes a mutation in one copy of the FGFR2 gene pair to cause the clinical signs of Apert syndrome. Apert syndrome is inherited in an autosomal dominant pattern of inheritance therefore if a person has Apert syndrome, his/her offspring would have a 50% risk of also having Apert syndrome.
Most children born with Apert syndrome are the first cases in their family. Their parents do not have the syndrome. This occurs because of a spontaneous mutation of the FGFR2 gene in the child. In such cases, the risk of having another child with Apert syndrome is very low. However, an increased risk for this condition has been found in pregnancies of older fathers.
What are the symptoms of Apert Syndrome?
- Craniosynostosis (premature closure of the sutures of the skull) causing the head to have a tall, “domed” appearance (also known as turribrachycephaly)
- Flat midface
- Wide set and bulging eyes
- Beaked nose
- Syndactyly of fingers and toes (fusion of the skin and bones of fingers and toes)
- Apert syndrome can also include various degrees of developmental delay, significant acne during the teen years, cleft palate, and brain anomalies
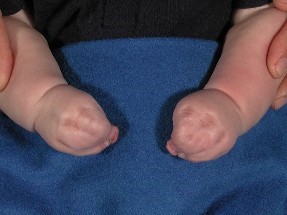
Photograph of syndactyly (fused fingers)
Photograph's of syndactyly (fused toes)
What are Apert Syndrome care options?
Multiple surgeries and ongoing therapies are required for these children but most will ultimately have useful function of the hands, attend school, have friends, and most of all enjoy life. With close follow-up by a craniofacial team, these children can grow to healthy and happy adults.
Apert Syndrome Treatments and Specialists
Apert syndrome is a lifelong condition with no cure. Your child will likely need to see several medical specialists and receive some of the following treatments throughout their care journey.
- Surgery. A child may require surgery shortly after birth if they have symptoms that affect the brain or skull. Other surgeries may be needed for the eyes, nose, chin, jaw, finger and toes.
- Hearing aids. Hearing specialists may need to prescribe hearing aids for children with hearing impairments.
- Therapy. Physical therapy, occupational therapy and speech therapy are all potential treatments for Apert syndrome.
- Breathing machines. If the child has airway obstruction, specialists may be needed to assess the breathing difficulties and prescribe a breathing machine.
- Dental care. Care of the child’s mouth and teeth is often critical for children with Apert syndrome.
- Vision care. Regular vision assessments and vision care are also a key part of Apert syndrome treatment.
This page was last updated on: December 15, 2023 04:45 PM