Pfeiffer Syndrome
Also known as: Acrocephalosyndactyly–Pfeiffer–type
What is Pfeiffer syndrome?
Pfeiffer syndrome is a genetic disorder characterized by early fusion of the bones of the skull (craniosynostosis). This in turn leads to abnormalities of the head and face. The disease is present at birth, and symptoms can persist and worsen as a person ages. The annual incidence of Pfeiffer syndrome is around one in every 100,000 births.
Pfeiffer syndrome was first described in 1964 by Dr. Rudolph Arthur Pfeiffer. This condition is also called acrocephalosyndactyly –Pfeiffer type which means “tall” head with variable fusion of the fingers.
What are the signs/symptoms of Pfeiffer Syndrome?
Pfeiffer syndrome has been divided into three clinical categories based on the clinical features found in the affected individual:
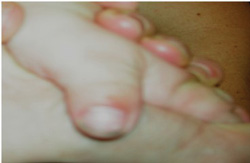
Type 1
- Craniosynostosis (premature closure of the skull sutures)
- Broad thumbs and great toes with variable fusion of bone/skin of fingers (syndactyly)
- Normal/near normal intelligence
Type 2
- Cloverleaf shaped skull due to craniosynostosis of multiple sutures.
- Severe ocular proptosis (protruding eyes)
- Broad thumbs and great toes
- Central nervous system anomalies
- Limited extension of elbow
Type 3
- Similar to type 2 without cloverleaf shaped skull
What causes Pfeiffer syndrome?
This condition is caused by a change (“mutation”) in a gene that is part of a group of genes called the Fibroblast Growth Factor Receptor Genes (FGFR). Mutations in two FGFR genes have been found in individuals with Pfeiffer syndrome. The two genes are FGFR1 and FGFR2.
Mutations in FGFR1 are usually found in the milder cases (Type 1) whereas mutations in FGFR2 are found in persons with Types 2 and 3. Pfeiffer syndrome is inherited in an autosomal dominant pattern of inheritance. Therefore, if a person has Pfeiffer syndrome, his/her offspring would have a 50 percent risk for also having Pfeiffer syndrome.
Most individuals with Pfeiffer syndrome are the first cases in the family. This means that the parents of the affected child do not have Pfeiffer syndrome. If a child’s parents do not have Pfeiffer syndrome, the risk to their other pregnancies is low.
How is Pfeiffer syndrome treated?
About 4 months after birth, children with Pfeiffer syndrome may require a surgery to relieve pressure on the skull (craniosynostosis) or fluid buildup (hydrocephalus). Then a shunt is inserted to continue to drain fluid away from the brain. Other treatments that may occur during the first year of life include opening the skull to allow the brain to grow and cosmetic or reconstructive surgery.
As the child grows, additional supportive treatments may be needed, including dental procedures, eye procedures and hearing assistive devices such as hearing aids.
What is the prognosis for children with Pfeiffer Syndrome?
With aggressive medical attention, the prognosis for most children with this condition is positive. Multiple surgeries and ongoing therapies are required for these children but most will ultimately, attend school, have friends, and most of all enjoy life. With close follow-up by a craniofacial team, most of these children can grow to be healthy adults.
There is no cure for Pfeiffer syndrome. The individual symptoms can be treated as they occur in order to give individuals with the disease the best quality of life possible. The Craniofacial team at Nicklaus Children’s Hospital has a large experience in treating patients with Pfeiffer Syndrome. Care is individualized for each child to maximize aesthetic and functional outcomes.
Reviewed by: Chad A Perlyn, MD
This page was last updated on: June 03, 2025 10:33 AM